戈登贝尔奖决赛选手用大型语言模型来预测新冠病毒变异株
2022-11-29 12:15
 分享到微信
分享到微信
 分享到微博
分享到微博
来自美国阿贡国家实验室、NVIDIA、芝加哥大学等组织机构的研究员开发了一个处理基因组规模数据的先进模型,并获得戈登贝尔 COVID-19 研究特别奖
这一戈登贝尔特别奖旨在表彰基于高性能计算的 COVID-19 研究。特别奖获得者教会了大型语言模型(LLMs)一种新的语言——基因序列,使这些模型能够提供基因组学、流行病学和蛋白质工程方面的洞察。
这项开创性的成果发表于 10 月,是由来自美国阿贡国家实验室、NVIDIA、芝加哥大学等组织机构的二十多名学术和商业研究员合作完成。
该研究团队训练了一个 LLM 来追踪基因突变,并预测需要关注的 SARS-CoV-2(导致 COVID-19 的病毒)变异株。虽然迄今为止大多数应用于生物学的 LLM 都是在小分子或蛋白质的数据集上训练的,但这一项目是在原始核苷酸序列(DNA 和 RNA 的最小单位)上训练的首批模型之一。
负责带领该项目的阿贡国家实验室计算生物学家 Arvind Ramanathan 表示:“我们假设从蛋白质水平到基因水平的数据有助于我们构建出更易于理解新冠病毒变异株的模型。通过训练模型去追踪整个基因组及其进化过程中的所有变化,我们不仅能够更好地预测 COVID,还能预测已掌握足够基因组数据的任何疾病。”
戈登贝尔奖被誉为 HPC 领域的诺贝尔奖。今年的戈登贝尔奖将在本周的 SC22 上由美国计算机协会颁发。该协会代表着全球约 10 万名计算领域的专家,自2020年开始向使用 HPC 推进 COVID-19 研究的杰出研究员颁发特别奖。
在一种只有四个字母的语言上
训练大型语言模型
长期以来,LLM 一直在接受人类语言的训练,这些语言通常由几十个字母组成,可以排列组合成数万个单词,并连接成长句和段落。而生物学语言只有四个代表核苷酸的字母,即 DNA 中的 A、T、G 和 C,或 RNA 中的 A、U、G 和 C。这些字母按不同顺序排列成基因。
虽然较少的字母看似会降低 AI 学习的难度,但实际上生物学语言模型要复杂得多。这是因为人类的基因组由超过 30 亿个核苷酸组成,而冠状病毒的基因组由大约 3 万个核苷酸组成,因此很难将基因组分解成不同、有意义的单位。
Ramanathan 表示:“在理解基因组这一‘生命代码’的过程中,我们所面对的一个主要挑战是基因组中的庞大测序信息。核苷酸序列的意义可能会受另一序列的影响,以人类的文本做类比,这种影响的范围不仅仅是文本中的下一句话或下一段话,而是相当于一本书中的整个章节。”
参与该项目协作的 NVIDIA 研究员设计了一种分层扩散方法,使 LLM 能够将约 1500 个核苷酸的长字符串当作句子来处理。
论文共同作者、NVIDIA AI 研究高级总监、加州理工学院计算+数学科学系布伦讲席教授 Anima Anandkumar 表示:“标准语言模型难以生成连贯的长序列,也难以学习不同变异株的基本分布。我们开发了一个在更高细节水平上运作的扩散模型,该模型使我们能够生成现实中的变异株,并采集到更完善的统计数据。”
预测需要关注的新冠病毒变异株
该团队首先使用细菌和病毒生物信息学资源中心的开源数据,对来自原核生物(像细菌一样的单细胞生物)超过 1.1 亿个基因序列进行了 LLM 预训练,然后使用 150 万个高质量的新冠病毒基因组序列,对该模型进行微调。
研究员还通过在更广泛的数据集上进行预训练,确保其模型能够在未来的项目中推广到其他预测任务,使其成为首批具备此能力的全基因组规模的模型之一。
在对 COVID 数据进行了微调后,LLM 就能够区分病毒变异株的基因组序列。它还能够生成自己的核苷酸序列,预测 COVID 基因组的潜在突变,这可以帮助科学家预测未来需要关注的变异株。
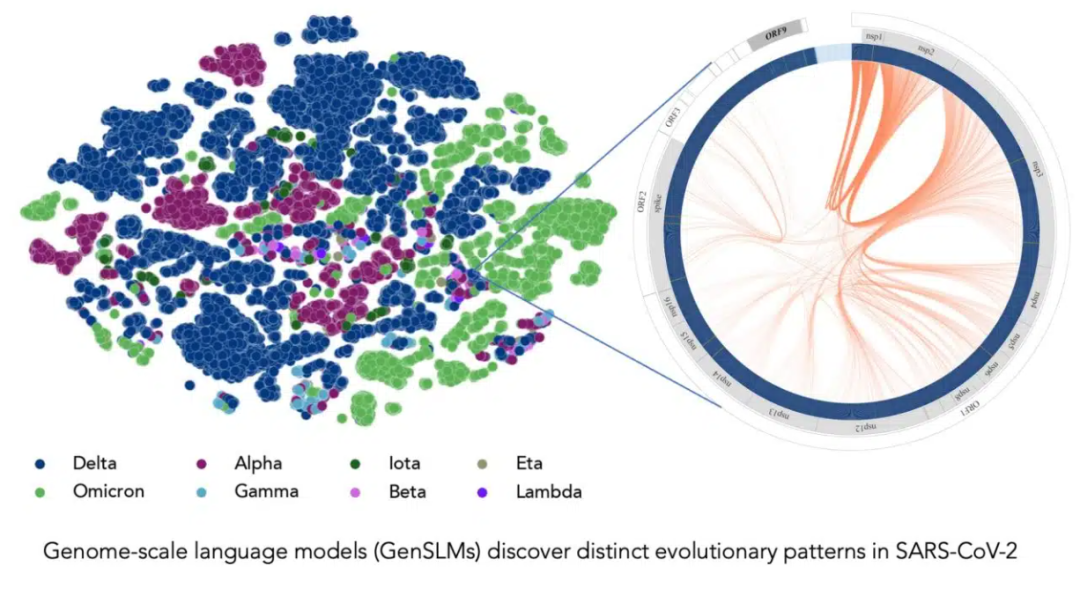
在长达一年时间内积累的 SARS-CoV-2 基因组数据的训练下,该模型可以推断出各种病毒株之间的区别。左边的每个点对应一个已测序的 SARS-CoV-2 病毒株,并按变异株颜色编码。右图放大了该病毒的一个特定毒株,它捕捉到了该毒株特有的病毒蛋白进化耦合关系。图片由美国阿贡国家实验室的 Bharat Kale、Max Zvyagin 和 Michael E. Papka 提供。
Ramanathan 表示:“大多数研究员一直在追踪新冠病毒突刺蛋白的突变,尤其是与人类细胞结合的域。但病毒基因组中还有其他蛋白质也会经历频繁的突变,所以了解这些蛋白质十分重要。”
论文中提到,该模型还可以与 AlphaFold、OpenFold 等常见的蛋白质结构预测模型整合,帮助研究员模拟病毒结构,研究基因突变如何影响病毒感染其宿主的能力。OpenFold 是 NVIDIA BioNeMo LLM 服务中包含的预训练语言模型之一。NVIDIA BioNeMo LLM 服务面向的是致力于将 LLM 应用于数字生物学和化学应用的开发者。
大幅加快 AI 训练速度
该团队在由 NVIDIA A100 Tensor Core GPU 驱动的超级计算机上开发 AI 模型,包括阿贡国家实验室的 Polaris、美国能源部的 Perlmutter 以及 NVIDIA 的 Selene 系统。通过扩展到这些强大的系统,他们在训练中实现了超过 1500 exaflops 的性能,创建了迄今为止最大的生物语言模型。
Ramanathan 表示:“我们如今处理的模型有多达 250 亿个参数,预计这一数量未来还会大幅增加。模型的尺寸、基因序列的长度、以及所需的训练数据量,都意味着我们的确需要搭载数千颗 GPU 的超级计算机来完成复杂的计算。”
研究员估计,训练一个具有 25 亿参数的模型版本,需要约 4000 个 GPU 耗时一个多月。该团队已经在研究用于生物学的 LLM,在公布论文和代码之前,他们在这个项目上已耗时约四个月。GitHub 页面上有供其他研究员在 Polaris 和 Perlmutter 上运行该模型的说明。
NVIDIA BioNeMo 框架可在 NVIDIA NGC 中心上的 GPU 优化软件中抢先体验。该框架将帮助研究员在多个 GPU 上扩展大型生物分子语言模型。作为 NVIDIA Clara Discovery 药物研发工具集的一部分,该框架将支持化学、蛋白质、DNA 和 RNA 数据格式。